Fanconi Anemia – Learn More About This Disease
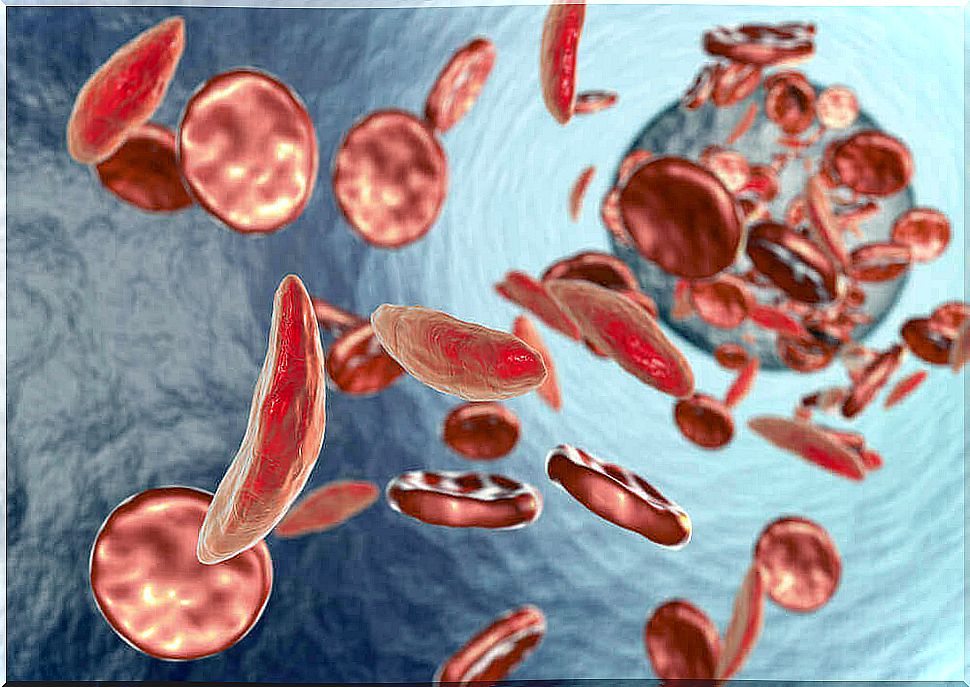
Fanconi’s anemia is an inherited disease that is passed on to children by their parents. It mainly affects the bone marrow and reduces the production of all types of blood cells. It is sometimes confused with Fanconi’s syndrome, but it is not the same.
Fanconi’s anemia mainly affects children. In 80% of cases, changes are found between the ages of 4 and 14, on average around the age of 7. However, cases have been reported in people from 0 to 35 years of age.
On average, one to five babies in a million newborns have Fanconi’s anemia. One in 300 people is able to transmit this disease. There are more cases in certain population groups. So far, the medical literature has described about 2,000 cases of this disease.
What is Fanconi anemia?
Fanconi anemia is an inherited disorder that causes bone marrow deficiency. As a result, the marrow does not produce enough blood cells, which translates into abnormalities in the functioning of the body.
This disorder impairs the bone marrow’s ability to produce blood cells such as red blood cells, white blood cells, and platelets. The former transport oxygen, the latter fight infection, and the third are responsible for blood clotting.
Sometimes, with Fanconi anemia, unusual cells are produced, which can cause serious diseases such as leukemia. Although this disease is a hematological disorder, i.e. a disorder involving blood, it can also affect other organs and systems.
Children with this condition are more likely to develop certain types of cancer and other serious diseases.
Causes and types of Fanconi anemia
This disorder is associated with the mutation of certain genes that are responsible for DNA repair and genome stability. For a child to inherit the disease, both parents must have abnormal genes.
So far, 13 genes have been identified as mutations that cause Fanconi anemia. These, in turn, are related to 8 dependent proteins. Therefore, this disease occurs in various forms with different course and characteristics.
For example, the FANC C variant has an earlier onset of symptoms and a lower survival. The FANC G subtype is associated with an increased risk of leukemia or tumors. The FANC D1, FANC J, and FANC N subtypes increase the risk of breast cancer.
Symptoms and diagnosis
The main symptom of this type of anemia is bone marrow failure. It causes a shortage of red and white blood cells and platelets. However, up to 40% of patients only have a platelet deficit, known as thrompocytopenia.
About 75% of patients with this disease show abnormalities from birth. Sometimes there are changes to the face, skin, hands or a very small structure. Anomalies also include internal organs such as the heart and kidneys.
It is estimated that up to 30% of patients also develop cancer. Tumors develop in 28% of patients. The diagnosis of the disease is based on clinical manifestations shown in tests such as:
- Morphology.
- Bone marrow biopsy.
- Study of chromosomal changes.
- X-ray of the hand.
- Audiometry and others.
Treatment
Most often, the treatment of Fanconi anemia is based on the transfusion of red blood cells and platelets, especially when symptoms are intense. Moreover, androgens are often prescribed to reduce anemia and avoid transfusions, but this can cause side effects.
Bone marrow transplantation is an effective method of disease control, but it does not rule out cancer. Hormone therapy with low doses of corticosteroids is an effective method for those who have not found a bone marrow donor.
People experiencing mild to moderate symptoms should get regular check-ups. Often, men with this disease have fertility problems. Survival rates are also highly variable.